
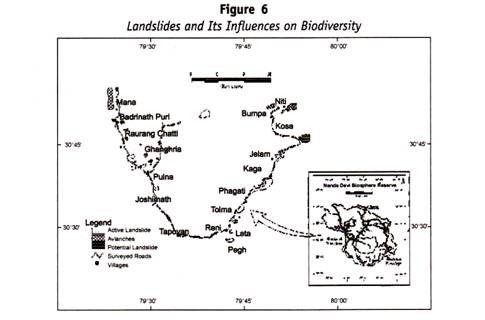
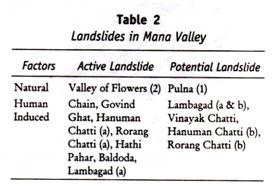
ADVERTISEMENTS:
Let us learn about Thin Layer Chromatography.
Introduction to Thin Layer Chromatography:
Chromatography is used to separate mixtures of substances into their components. All forms of chromatography work on the same principle. They all have a stationary phase (a solid, or a liquid supported on a solid) and a mobile phase (a liquid or a gas).
The mobile phase flows through the stationary phase and carries the components of the mixture with it. Different components travel at different rates. Thin layer chromatography is done exactly as it says-using a thin, uniform layer of silica gel or alumina coated onto a piece of glass, metal or rigid plastic.
ADVERTISEMENTS:
The silica gel (or the alumina) is the stationary phase. The stationary phase for thin layer chromatography also often contains is substance which fluoresces in UV light. The mobile phase is a suitable liquid solvent or mixture of solvents.
Producing the Chromatogram:
The chromatogram can be produced by dipping the chromatographic plate (TLC plate with silica and mixture of different spots to be separated) in the mixture of various solvent systems (for e.g., n-Butanol, Glacial Acetic Acid and Distilled Water) in a specific ratio for separating amino acids using standard amino acid kit with known Rf values.
A pencil line is drawn near the bottom of the plate Fig. 22.2 and a small drop of solution of the dye mixture is placed on it. Any labeling on the plate to show the original position of the drop must also be in pencil. If any of this was done in ink, dyes from the ink would also move as the chromatogram developed.
When the spot of mixture is dry, the plate is stood in a shallow layer of solvent in a covered beaker, it is important that the solvent level is below the line with the spot on it.

The reason of covering the beaker is to make sure that the atmosphere in the beaker or the chromatographic chamber is saturated with solvent vapour. To help this, the beaker is often lined with some filter paper soaked in the solvent. Saturating the atmosphere in the beaker with vapour stops the solvent from evaporating as it rises up the plate.
As the solvent slowly travels up the plate, the different components of the dye mixture travel at different rates and the mixture is separated into different spots.
The diagram shows the plate after the solvent has moved about half way up it Fig. 22.10.

The solvent is allowed to rise until it almost reaches the top of the plate fig. 22.11.

That will give the maximum separation of the dye components for this particular combination of solvent and stationary phase.
Measuring Rf Values:
If all you wanted to know is how many different dyes made up the mixture, you could just stop there. However, measurements are often taken from the plate in order to help identify the compounds present. These measurements are the distance traveled by the solvent, and the distance traveled by individual spots.
When the solvent front gets close to the top of the plate, the plate is removed from the beaker and the position of the solvent is marked with another line before it has a chance to evaporate.
ADVERTISEMENTS:
These measurements are then taken:
The Revalue for each dye is then worked out using the formula:
Rf = distance travelled by component /distance travelled by solvent
For example, if the one component travelled 170mm from the base line while the solvent had traveled 50mm, then the Rf value for that dye is:
ADVERTISEMENTS:
If you could repeat this experiment under exactly the same conditions, then the Rf value for each dye would always be the same. For example, the Rf value for the above dye would always be 3.4. However if anything changes (the temperature, the exact composition of the solvent, and so on), that is no longer true.
What if the substances you are interested in all colourless ?
There are two simple ways of getting around this problem:
Using Fluorescence:
In case of mixtures of generating colourless spots in stationary phase on a thin layer plate often a substance is added to it which will fluoresce when exposed to UV light. That means that if you shine UV light on it, will glow fig. 22.12.

That glow is masked at the position where the spots are on the final chromatogram-even if those spots are invisible to eye. That means that if you shine UV light on the place, it will all glow apart from where the spots are. The spots show up as darker patches.
While the UV is still shining on the plate you obviously have to mark the position of spots by
drawing a pencil circle around then. As soon as you switch off the UV source, the spots will disappears again.
Showing the Spots Up Chemically:
In some cases, it may be possible to make the spots visible by reacting them with something which produces a coloured product. A good example of this is in chromatograms produces from amino acids mixtures Fig. 22.13.

The chromatogram is air-dried and is then sprayed with a solution of ninhydrin and later allowed to dry (at about 100° C in oven for few minutes). Ninhydrin reacts with amino acids to give coloured compounds, mainly brown or purple.
Using Thin Layer Chromatography to Identify Compounds:
Suppose you had a mixture of amino acids and wanted to find out which particular acids the mixture contained. For simplicity we, will assume that you know the mixture can only possibly contain five of the amino acids.
A small drops of the mixture is placed on the base line of the thin layer plate, and similar small spots of the known amino acids are placed along-side it. The plate is then stood in a suitable solvent and left to develop as before. In the diagram the mixture is M, and the known amino acids are labeled 1 to 5.
The left hand diagram shows the plate after the solvent front has almost reached the top. The spots are still invisible. The second diagram shows what is might look like after spraying with ninhydrin fig. 22.13 and 22.14.

There is no need to measure to Rf values because you can easily compare the spots in the mixture with those of the known amino acids-both from their positions and their colours.
How does thin layer Chromatography Work?
The Stationary Phase-Silica Gel:
ADVERTISEMENTS:
Silica gel is a form of silicon dioxide (silica). The silicon atoms are joined via oxygen atoms in a giant covalent structure. However, at the surface of the silica gel, the silicon atoms are attached to —OH groups.
So, at the surface of the silica gel you have Si-O-H bonds instead of Si-O-Si bonds. The diagram shows a small part of the silica surface.

The surface of the silica gel is very polar and, because of the-OH groups, can from hydrogen bonds with suitable compounds around it as well as vander Walls dispersion forces and dipole attractions.
The other commonly used stationary phase is alumina-aluminium oxide. The aluminium atoms on the surface of this also have -OH groups attached. Anything we say about silica gel therefore applies equally to alumina.
Separation of the Compounds in a Developing Chromatogram:
As the solvent beings to soak up the plate, it first dissolves the compounds in the spot that you have put on the base line. The compounds present with then tend to get carried up the chromatography plate as the solvent continues to move upwards.
ADVERTISEMENTS:
How fast the compounds get carried up the plate depends on two things:
1. How soluble the compound is in the solvent. This will depend on how much attraction there is between the molecules of the compound and those of the solvent.
2. How much the compound sticks to the stationary phase the silica get, for example. This will depend on how much attraction there is between the compound and the silica gel.
Suppose the original spot contained two compounds-one of which can from hydrogen bonds, and one of which can only take part in weaker vander Walls interactions, the one which can hydrogen bond will stick to the surface of the silica gel more firmly then the other one. We say that one the adsorbed more strongly than the other.
Adsorption is the name given to one substance forming some sort of bonds to the surface of another one. Adsorption is not permanent-there is a constant movement of a molecule between being adsorbed on to the silica gel surface and going back into solution in the solvent.
Obviously the compound can only travel up the plate during the time that it is dissolved in the solvent. While it is adsorbed on the silica gel, it is temporarily stopped-the solvent is moving on without it. That means that the more strongly a compound is adsorbed, the less distance it can travel up the plate.
In the example we started with, the compound which can hydrogen bond will adsorb more strongly than the one dependent on van der Walls interactions, and so won’t travel so far up the plate. What if both components of the mixture can hydrogen bond?
It is very unlikely that bond will hydrogen bond to exactly the same extent, and be soluble in the solvent to exactly the same extent. It isn’t just the attraction of the compound for the silica gel which matters. Attractions between the compound and the solvent are also important-they will affect how easily the compound is pulled back into solution away from the surface of the silica.
Few methods of chemical analysis are truly specific to a particular analyte. It is often found that the analyte of interest must be separated from the myriad of individual compounds that may be present in a sample. As well as providing the analytical scientist with methods of separation, chromatographic technique can also provide methods of analysis.
Chromatography involves a sample (or sample extract) being dissolved in a mobile phase (which may be a gas, a liquid or a supercritical fluid). The mobile phase is then forced through an immobile, immiscible stationary phase. The phases are chosen such that components of the sample have differing solubilities in each phase.
A component which is quite soluble in the stationary phase will take longer to travel through it than a component which is not very soluble in the stationary phase but very soluble in the mobile phase. As a result of these differences in mobilities, sample components will become separated from each other as they travel through the stationary phase.
Techniques such as H.P. L.C. (High Performance Liquid Chromatography) and G.C. (Gas Chromatography) use columns-narrow tubes packed with stationary phase, through which the mobile phase is forced. The sample is transported through the column by continuous addition of mobile phase.
This process is called elution. The average rate at which an analyte moves through the column is determined by the time it spends in the mobile phase.
Distribution of Analytes between Phases:
The distribution of analytes between phases can often be described quite simply. An analyte is in equilibrium between the two phases;

The equilibrium constant, K, is termed the partition coefficient; defined as the molar concentration of analyte in the stationary phase divided by the molar concentration of the analyte in the mobile phase.
The time between sample injection and an analyte peak reaching a detector at the end of the column is termed the retention time (tR). Each analyte in a sample will have a different retention time. The time taken for the mobile phase to pass through the column is called fM.
 and Retentio factor (tM)")
A term called the retention factor, K is often used to describe the migration rate of an analyte on a column.
It is also called the capacity factor. The retention factor for analyte A is defined as:
K’A = tR − tM / tM
tR and tM are easily obtained from a chromatogram. When an analytes retention factor is less than one, elution is so fast that accurate determination of the retention time is very difficult. High retention factors (greater that 20) mean that elution takes a very long time. Ideally, the retention factor for an analyte is between one and five.
We define a quantity called the selectivity factor, α , which describes the separation of two species (A and B) on the column;
α = k’B /k’A
When calculating the selectivity factor, species A elutes faster the species B. The selectivity factor is always greater than one.
The Theoretical Plate Model of Chromatography:
The plate model supposes that the chromatographic column is contains a large number of separate layers, called theoretical plates Fig. 22.16. Separate equilibrations of the sample between the stationary and mobile phase occur in these “plates”. The analyte moves down the column by transfer of equilibrated mobile phase from one plate to the next.
It is important to remember that the plates do not really exist; they are a figment of the imagination that helps us understand the processes at work in the column. They also serve as a way of measuring column efficiency, either by stating the number of theoretical plates in a column. N (the more plates the better), or by stating the plate height; Height Equivalent to a Theoretical Plate (the smaller the better).
If the length of the column is L, then the HETP is:
HETP = L / N
The number of theoretical plates that a real column possesses can be found by examining a chromatographic peak after elution;

where w1/2 is the peak width at half-height.
As can be seen from this equation, columns behave as if they have different number of plates for different solutes in a mixture.
The Rate Theory of Chromatography:
A more realistic description of the processes at work inside a column takes account of the time taken for the solute to equilibrate between the stationary and mobile phase (unlike the plate model, which assumes that equilibration is infinitely fast).
The resulting band shape of a chromatographic peak is therefore affected by the rate of elution. It is also affected by the different paths available to solute molecules as they travel between particles of stationary phase. If we consider the various mechanisms which contribute to band broadening, we arrive at the Van Deemter equation for plate height.
HETP = A + B / u + Cu
where u is the average velocity of the mobile phase. A, B, and C are factors which contribute to band broadening.
A-Eddy Diffusion:
The mobile phase moves through the column which is packed with stationary phase. Solute molecules will take different paths through the stationary phase at random. This will cause broadening of the solute band, because different paths are of different lengths.
B-Longitudinal Diffusion:
The concentration of analyte is less at the edges of the band than at the center. Analyte diffuses out from the center to the edges. This causes band broadening. If the velocity of the mobile phase in high then the analyte spends less time on the column, which decreases the effects of longitudinal diffusion.
C-Resistance to Mass Transfer:
The analyte takes a certain amount of time to equilibrate between the stationary and mobile phase. If the velocity of the mobile phase is high, and the anlyte has a strong affinity for the stationary phase, then the analyte in the mobile phase will move ahead of the analyte in the stationary phase. The bond of analyte is broadened. The higher the velocity of mobile phase, the worse the broadening becomes.
Van Deemter Plots:
A plot of plate height vs. average linear velocity of mobile phase is called Van Deemter Plot (Fig. 22.17).

Such plots are of considerable use in determining the optimum mobile phase flow rate.
Resolution:
Although the selectivity factor, α , describes the separation of band centres, it does not take into account peak widths. Another measure of how well species have been separated is provided by measurement of the resolution.
The resolution of two species, A and B, is defined as:
Baseline resolution is achieved when R= 1.5
It is useful to relate the resolution to the number of plates in the column, the selectivity factor and the retention factors of the two solutes:

To obtain high resolution, the three terms must be maximized. An increase in N, the number of theoretical plates, by lengthening the column leads to an increase in the retention time and increased band broadening-which may not be desirable. Instead, to increase the number of plates, the high equivalent to a theoretical plate can be reduced by reducing the size of the stationary phase particles.
It is often found that by controlling the capacity factor, k’ separations can be greatly improved. This can be achieved by changing the temperature. (In Gas Chromatography) or the composition of the mobile phase (in Liquid Chromatography)
The selectivity factor, ∝, can also be manipulated to improve separation. When ∝ is close to unity, optimizing k’ and increasing N is not sufficient to give good separation in a reasonable time.
In these case, K’ is optimized 1st, and then ∝ is increased by one of the following procedure:
1. Changing mobile phase composition.
2. Changing column temperature.
3. Changing composition of stationary phase.
4. Using special chemical effects. (Such as incorporating a spices which complexes with one of the solutes into the stationary phase).