ADVERTISEMENTS:
The following points highlight the top seven processes helping amino acids to form succinyl-CoA. The processes are: 1. Methionine 2. Trans-methylation 3. Leucine 4. Valine 5. Isoleucine 6. Catabolism of Histidine 7. Metabolism of Glycine.
Amino Acids Forming Succinyl-CoA: Process # 1. Methionine:
a. Methionine is first activated by ATP forming S-adenosylmethionine (“active methionine”).
b. After removal of the methyl group, the active methionine is converted into S-adenosyl-homocysteine. This is hydrolyzed to produce homocysteine and adenosine.
ADVERTISEMENTS:
c. Homocysteine then condenses with a molecule of serine forming cystathionine which produces homoserine and cysteine on hydrolytic cleavage.
d. Homoserine is then converted to α-ketobutyric acid by the enzyme homoserine deaminase.
e. The oxidative decarboxylation converts α-ketobutyrate to propionyl-CoA.
f. Propionyl-CoA is converted to D-methyl malonyl-CoA by propionyl CoA carboxylase in presence of ATP and biotin as Coenzyme.
ADVERTISEMENTS:
g. Methyl-malonyl-CoA reeemase converts D-methyl malonyl-CoA to L-methyl-malonyl-CoA which in turn, is converted into succinyl-CoA by the specific isomerase with B12 as coenzyme.
The reactions are given (Fig. 20.39).

Amino Acids Forming Succinyl-CoA: Process # 2. Trans-methylation:
a. The transfer of CH3 group from a compound of the body to a suitable acceptor which has no CH3 group. Such a reaction is said to be trans-methylation reaction.
b. The CH3 donor is biologically labile -CH3 group.
The most important compounds with biologically labile methyl groups are:
(i) Active methionine containing -S-CH, group.
ADVERTISEMENTS:
(ii) Choline containing N+ CH3CH3CH3.
(iii) Betaine, an oxidative derivative of choline.
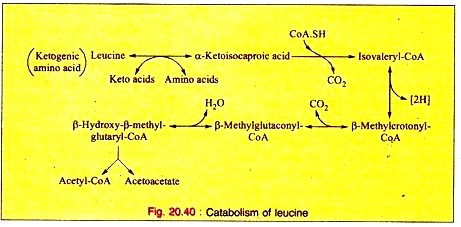
Amino Acids Forming Succinyl-CoA: Process # 3. Leucine:
a. Leucine is trans-aminated to produce α-ketoisocaproic acid, which, on decarboxylation, yields isovaleryl-CoA. This on dehydrogenation produces β-methyl-crotonyl-CoA.
b. Carboxylation converts (3-methylcrotonyl-CoA into β-methyl-glutaconyl-CoA which in turn, is converted to β-hydroxy- β-methyl-glutaryl-CoA on hydration. This product is cleaved into acetoacetate plus acetyl-CoA.
ADVERTISEMENTS:
The reactions are given below (Fig. 20.40).
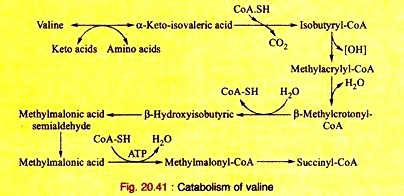
Amino Acids Forming Succinyl-CoA: Process # 4. Valine:
a. Valine, on transamination, produces α-ketoisovaleric acid which, on decarboxylation yields isobutyryl-CoA. This, on dehydrogenation, produces methyl-acrylyl-CoA.
b. Methyl-acrylyl-CoA is hydrated to produce β-hydroxyisobutyryl-CoA which, on hydrolysis, gives β-hydroxyisobutyric acid. This is converted into methylmalonic acid semi-aldehyde.
ADVERTISEMENTS:
c. The semi-aldehyde is oxidized to yield methyl-malonic acid which, on acylation, yields methyl-malonyl-CoA. This ultimately produces succinyl-CoA as shown in leucine catabolism.
The reactions are given in Fig 20.41.
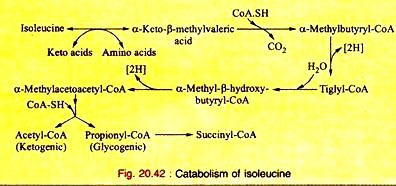
Amino Acids Forming Succinyl-CoA: Process # 5. Isoleucine:
a. Isoleucine, on transamination, produces α-keto-β-methyl-valeric acid which, on decarboxylation, yields α-methyl-butyryl- CoA. This, on dehydrogenation, produces tiglyl-CoA.
b. Tiglyl-CoA is hydrated to produce α-methyl-β-hydroxybutyryl-CoA which, on dehydrogenation, forms α-methyl-acetoacetyl-CoA. Thiolytic cleavage forms acetyl-CoA plus propionyl-CoA. Propionyl-CoA is converted to succinyl- CoA as mentioned in leucine catabolism.
Amino Acids Forming Succinyl-CoA: Process # 6. Catabolism of Histidine:
The catabolism of histidine proceeds under the following pathways:
a. Urocanic Acid Pathway.
b. Histamine Pathway.
c. Imidazole Pyruvic Acid Pathway.
d. Minor Pathways.
ADVERTISEMENTS:
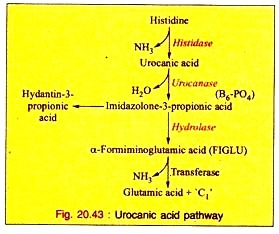
a. Urocanic Acid Pathway:
This is the major pathway of histidine catabolism:
i. Histidine is first converted into urocanic acid by the removal of one mol of ammonia by the enzyme histidase.
ii. The enzyme urocanase with pyridoxal phosphate (B6-PO4) as coenzyme converts urocanic acid to imidazolone-3-propionic acid which, in turn, is converted into α-formiminoglutamic acid (FIGLU) by hydrolase.
iii. α-Formimino glutamic acid is then converted into glutamic acid, resulting in the transfer of C1 carbon atom to tetrahydrofolic acid.
In human beings and animals, deficiency of folic acid or vitamin Bi: causes an excessive excretion of FIGLU. A small amount of imidazole propionic acid may be oxidized to hydantin-3-propionic acid which is excreted in the urine. The reactions are stated below (Fig. 20.43).
b. Histamine Pathway:
Histidine is decarboxylated by histidine decarboxylase to form histamine. This is formed at a significant rate in fetal and regenerating liver as well as in healing skin wounds. Histamine is detoxified as its methyl or acetyl derivatives or to oxidize to imidazole acetate which are excreted in urine. The reactions are given above (Fig. 20.44).

c. Imidazole Pyruvic Acid Pathway:
Histidine, by transamination, is converted into imidazole pyruvic acid which is further oxidized to imidazole acetic acid as:

d. Minor Pathways:
(a) Histidine is converted into carnosine and aniserine by minor pathways. These compounds are found in muscle. Carnosine is a peptide of histidine and β-alanine; whereas aniserine is a peptide of methyl-histidine and β-alanine.
2. Ergothionine, the betaine of 2-mercaptohistidine, is present in high concentration in human erythrocytes. It is also found in liver and brain.
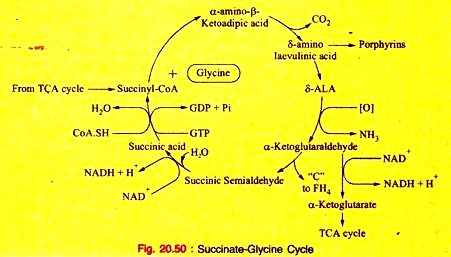
Amino Acids Forming Succinyl-CoA: Process # 7. Metabolism of Glycine:
Glycine is non-essential, but it is an important amino acid since it produces significant biological compounds in the body.
a. Deamination:
The enzyme glycine oxidase (a flavo-protein enzyme) available in liver and kidney deaminates glycine to produce glyoxalate (see Fig. 20.45).
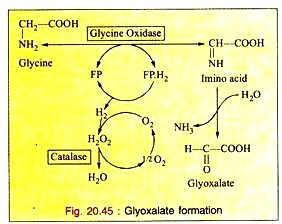
Glyoxalate is further converted to either oxalic acid or formic acid which enters “One carbon Pool”.
b. Formation of CO2, NH3 and N5, N10-Methyiene FH4:
Glycine is catabolized by the enzyme glycine synthase complex, found as aggregate in mitochondria, to CO2, NH3 and N5, N10– methylene FH4. This reversible reaction occurs in liver of most vertebrates including humans (Fig. 20.46).
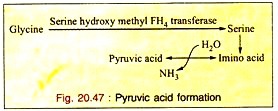
c. Formation of Pyruvic Acid through Serine:
Glycine is first converted to serine which is converted into pyruvic acid by non-oxidative-deamination. So glycine is a glycogenic amino acid (Fig. 20.47).
d. Formation of Amino Acetone:
Glycine is oxidized to amino acetone by the enzyme amino-acetone synthetase in presence of acetyl-CoA. The amino-acetone is further metabolized to lactic acid and pyruvic acid via methyl glyoxal (Fig. 20.48).

Utilization of Glycine:
a. Glycine is necessary for the synthesis of heme.
b. Glycine molecule as a whole is used to form C4, C5, and N7 of Purine nucleus.
c. It is required in the formation of glutathione, a tripeptide, consisting of other amino acids, cysteine and glutamic acid in addition to Glycine.
d. The enzyme transamidinase present in kidney catalyzes the conversion of glycine and arginine to Glycocyamine (Guanido acetic acid) which is further converted to creatine in the liver.
e. It acts as a conjugating agent. Glycine conjugates benzoic acid and other aromatic acid, produced during the processes of metabolism to form a conjugated compound before excreted in urine.
(a)
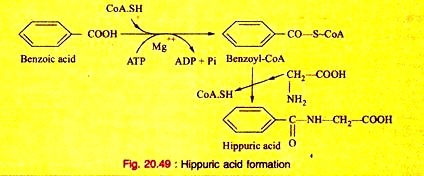

f. Glycine is converted to pyruvic acid. Hence, it is glycogenic.
g. Glycine is a source of format and oxalate.
Inherited Disorder of Glycine Metabolism:
a. Glycinuria – See in inborn errors of Protein metabolism.
b. Primary hyperoxaluria in inborn errors of Protein metabolism (Fig. 20.50).